Electronic and Topological Properties of UIO66-ILs Interaction through a Computational Study
DOI:
https://doi.org/10.37934/aram.102.1.4453Keywords:
Binding energies, molecular docking, metal-organic framework, ionic liquid, adsorbentAbstract
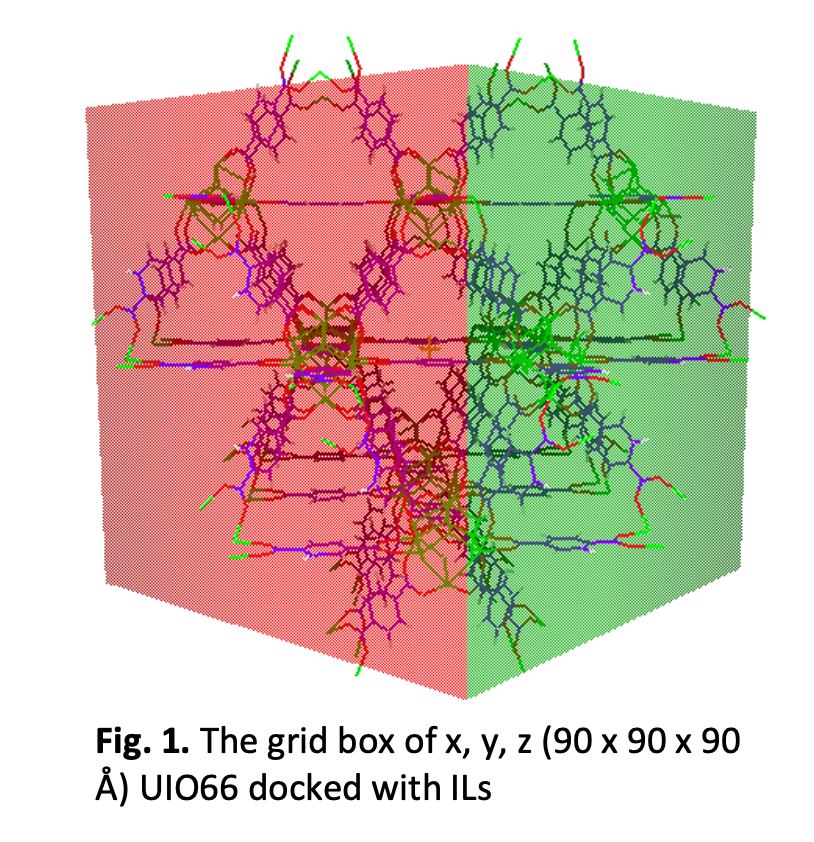
A high surface area adsorbent, such as MOFs, is an attractive option to evacuate heavy metals from wastewater. However, due to the possibility of the MOF structure collapsing when exposed to water, the stability and selectivity of MOFs for wastewater treatment remain a debatable task. Depending on the application, it has been shown that the consolidation of IL into MOF could improve the stability and reusability of pure MOF; nevertheless, research on the molecular interactions between the MOF framework and IL is still lacking. The stability, compatibility, and selectivity of this hybrid material are significantly influenced by the fundamental interactions between the MOFs and ILs moieties (cation and anion molecules), particularly the sorts of interactions. In this work, we focused on the topological properties of ILs and utilization of molecular docking calculations to assess the binding energy (ΔGbind) and binding affinity between UIO66 with TFSI-imidazole and TFSI-pyridinium ILs studied. The properties of ILs such as aromaticity of the structures, bond critical point (BCP), and electrostatic potential map surface were carried out using the Gaussian and Multiwfn software while the binding energies were computed using the AutoDock software to identify particular binding sites for IL toward MOF. The Autodock software's default setting was used to prepare all of the structures. To determine the binding locations of MOF, the IL was successively incorporated into the MOF pores inside the grid box of 90 x 90 x 90 Å (x, y, and z) dimensions. The results showed that both ILs; TFSI-imidazole, and TFSI-pyridinium IL, interacted favorably with the MOFs, having binding energies varying from -4.12 to -8.60 kcal mol-1. The stability of UIO66-ILs system increases with the addition of anion and cation. According to docking molecular structures, both cation and anion tend to bind in the corner of the MOF pore, which is in line with the previous work.
Downloads